Protein contact map
A protein contact map represents the distance between all possible residue pairs of a three-dimensional protein structure using a binary two-dimensional matrix. For two residues 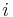 and
and 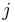 , the
, the 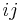 element of the matrix is 1 if the two residues are closer than a predetermined threshold, and 0 otherwise. Various contact definitions have been proposed: The distance between the Cα-Cα atom with threshold 6-12 Å; distance between Cβ-Cβ atoms with threshold 6-12 Å (Cα is used for Glycine); distance between the side-chain centers of mass; distance between the Van der Waals radius of selected atoms; or consideration of any atom and lower threshold 4.5-6 Å.
element of the matrix is 1 if the two residues are closer than a predetermined threshold, and 0 otherwise. Various contact definitions have been proposed: The distance between the Cα-Cα atom with threshold 6-12 Å; distance between Cβ-Cβ atoms with threshold 6-12 Å (Cα is used for Glycine); distance between the side-chain centers of mass; distance between the Van der Waals radius of selected atoms; or consideration of any atom and lower threshold 4.5-6 Å.
Contact maps provide a more reduced representation of a protein structure than its full 3D atomic coordinates. The advantage is that contact maps are invariant to rotations and translations. They are more easily predicted by machine learning methods. It has also been shown that under certain circumstances it is possible to reconstruct the 3D coordinates of a protein using its contact map.[1]
Contact maps are also used for protein superimposition and to describe similarity between protein structures.[2]. They are either predicted from protein sequence or calculated from a given structure.
Contact maps have been used in the prediction of membrane proteins where helix-helix interactions are targeted.[3]
See also
References
- ^ Vassura M, Margara L, Di Lena P, Medri F, Fariselli P, Casadio R (2008). "Reconstruction of 3D Structures From Protein Contact Maps". IEEE/ACM Transactions on Computational Biology and Bioinformatics 5 (3): 357–367. doi:10.1109/TCBB.2008.27. PMID 18670040.
- ^ Holm L, Sander C (August 1996). "Mapping the protein universe". Science 273 (5275): 595–603. doi:10.1126/science.273.5275.595. PMID 8662544.
- ^ Lo A, Chiu YY, Rødland EA, Lyu PC, Sung TY, Hsu WL. (2009). "Predicting helix-helix interactions from residue contacts in membrane proteins". Bioinformatics 25 (8): 996–1003. doi:10.1093/bioinformatics/btp114. PMC 2666818. PMID 19244388. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2666818.
External links
- GPCPRED — a web based contact prediction server
- DISTILL — prediction of protein structural features (including protein residue contact maps)
- Structural Proteomics Tools — includes amino acid contact maps
- ProfCon — prediction of inter-residue contacts
- TMHcon — prediction of helix-helix contacts specifically within the transmembrane parts of membrane proteins
- TMhit — A new transmembrane helix-helix interaction prediction method based on residue contacts
- CMAPpro — A protein contact map prediction server